Decorate LFQData with Methods for transforming Intensities
Source:R/LFQDataTransformer.R
LFQDataTransformer.RdDecorate LFQData with Methods for transforming Intensities
Decorate LFQData with Methods for transforming Intensities
Methods
Method log2()
log2 transform data
Method robscale()
robust scale data
Method robscale_subset()
log2 transform and robust scale data based on subset
Usage
LFQDataTransformer$robscale_subset(
lfqsubset,
preserve_mean = TRUE,
colname = "transformed_abundance"
)Method intensity_array()
Transforms intensities
Method intensity_matrix()
pass a function which works with matrices, e.g., vsn::justvsn
Examples
istar <- sim_lfq_data_peptide_config(Nprot = 50)
#> creating sampleName from file_name column
#> completing cases
#> completing cases done
#> setup done
lfqdata <- LFQData$new(istar$data, istar$config)
lfqcopy <- lfqdata$get_copy()
lfqTrans <- lfqcopy$get_Transformer()
x <- lfqTrans$intensity_array(log2)
#> Column added : log2_abundance
x$lfq$is_transformed()
#> [1] TRUE
x <- x$intensity_matrix(robust_scale)
#> Warning: data already transformed. If you still want to log2 tranform, set force = TRUE
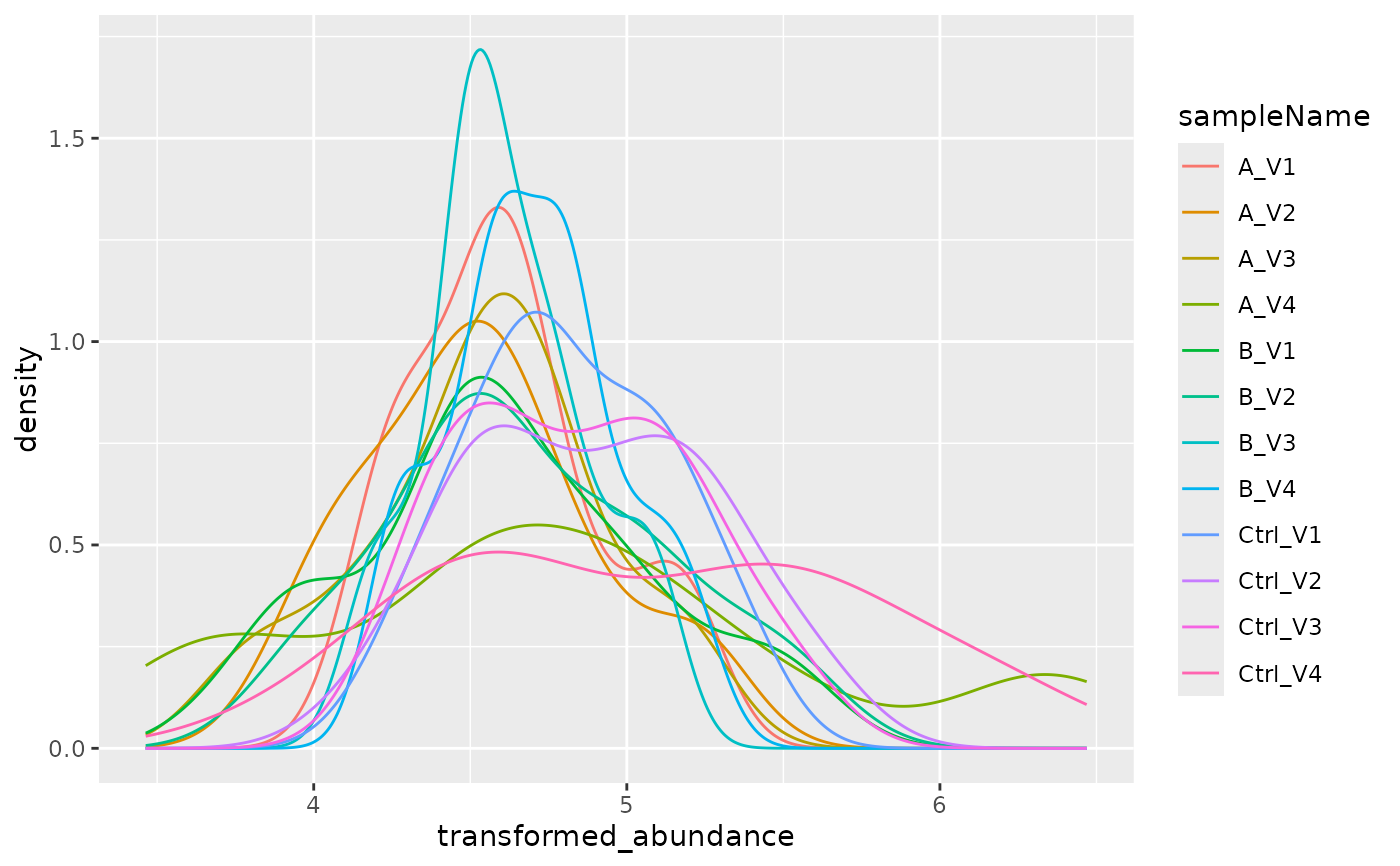
plotter <- x$lfq$get_Plotter()
plotter$intensity_distribution_density()
#> Warning: Removed 225 rows containing non-finite outside the scale range
#> (`stat_density()`).
 # transform by asinh root and scale
lfqcopy <- lfqdata$get_copy()
lfqTrans <- lfqcopy$get_Transformer()
x <- lfqTrans$intensity_array(asinh)
#> Column added : asinh_abundance
mads1 <- mean(x$get_scales()$mads)
x <- lfqTrans$intensity_matrix(robust_scale, force = TRUE)
#> Joining with `by = join_by(sampleName, isotopeLabel, protein_Id, peptide_Id)`
mads2 <- mean(x$get_scales()$mads)
stopifnot(abs(mads1 - mads2) < 1e-8)
stopifnot(abs(mean(x$get_scales()$medians)) < 1e-8)
lfqcopy <- lfqdata$get_copy()
lfqTrans <- lfqcopy$get_Transformer()
lfqTrans$log2()
#> Column added : log2_abundance
before <- lfqTrans$get_scales()
lfqTrans$robscale()
#> data is : TRUE
#> Joining with `by = join_by(sampleName, isotopeLabel, protein_Id, peptide_Id)`
after <- lfqTrans$get_scales()
stopifnot(abs(mean(before$medians) - mean(after$medians)) < 1e-8)
stopifnot(abs(mean(before$mads) - mean(after$mads)) < 1e-8)
# normalize data using vsn
lfqcopy <- lfqdata$get_copy()
lfqTrans <- lfqcopy$get_Transformer()
lfqTransCheck <- lfqcopy$get_Transformer()
lfqTransCheck$log2()
#> Column added : log2_abundance
lfqTransCheck$get_scales()
#> $medians
#> A_V1 A_V2 A_V3 A_V4 B_V1 B_V2 B_V3 B_V4
#> 4.463740 4.450835 4.444549 4.463231 4.537348 4.568077 4.495236 4.519219
#> Ctrl_V1 Ctrl_V2 Ctrl_V3 Ctrl_V4
#> 4.500299 4.453443 4.491344 4.464994
#>
#> $mads
#> A_V1 A_V2 A_V3 A_V4 B_V1 B_V2 B_V3 B_V4
#> 0.3515227 0.3699336 0.3679643 0.3853720 0.3884622 0.3747087 0.3602410 0.3672785
#> Ctrl_V1 Ctrl_V2 Ctrl_V3 Ctrl_V4
#> 0.3507487 0.3606693 0.3632381 0.3691903
#>
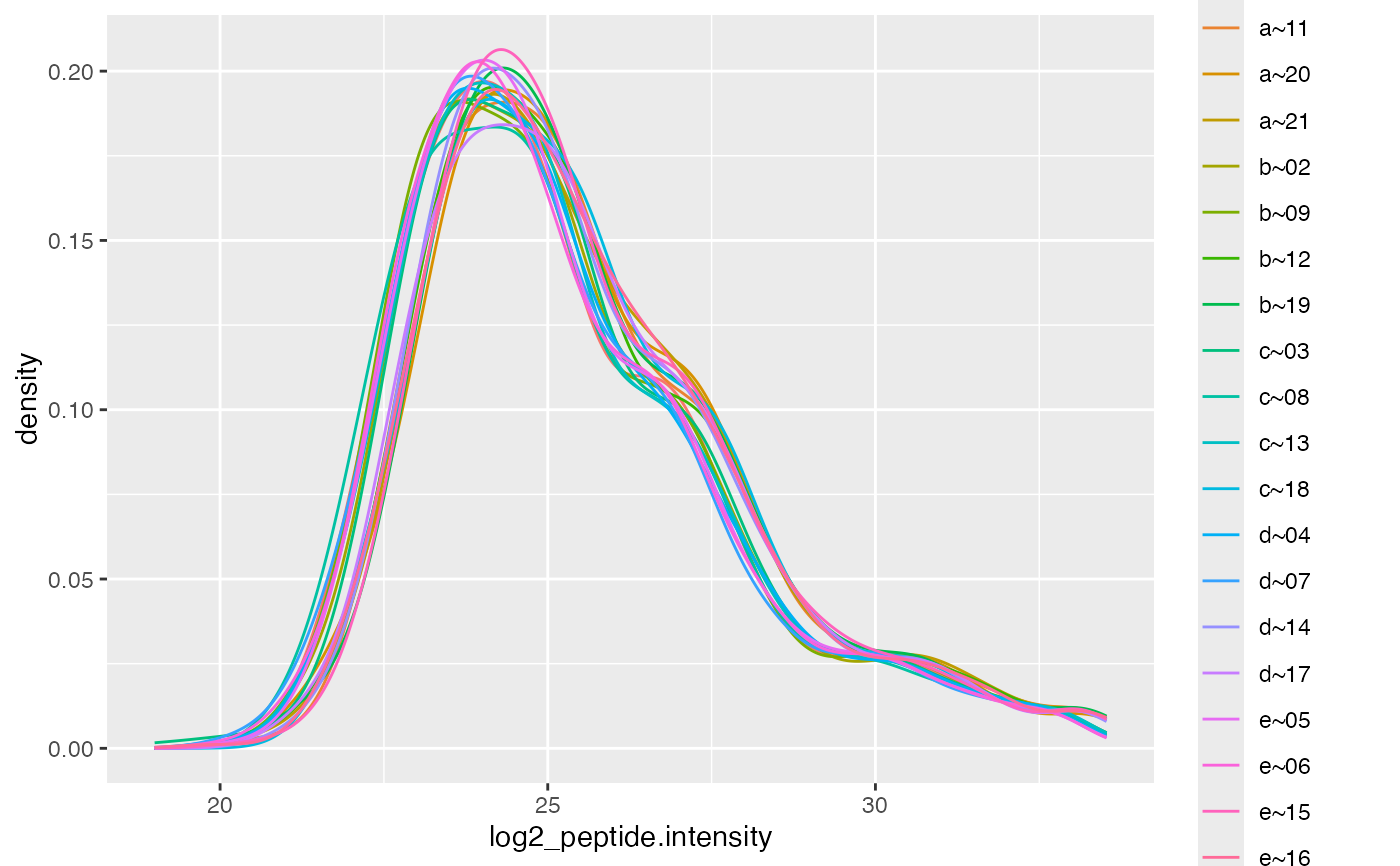
lfqTransCheck$lfq$get_Plotter()$intensity_distribution_density()
#> Warning: Removed 225 rows containing non-finite outside the scale range
#> (`stat_density()`).
# transform by asinh root and scale
lfqcopy <- lfqdata$get_copy()
lfqTrans <- lfqcopy$get_Transformer()
x <- lfqTrans$intensity_array(asinh)
#> Column added : asinh_abundance
mads1 <- mean(x$get_scales()$mads)
x <- lfqTrans$intensity_matrix(robust_scale, force = TRUE)
#> Joining with `by = join_by(sampleName, isotopeLabel, protein_Id, peptide_Id)`
mads2 <- mean(x$get_scales()$mads)
stopifnot(abs(mads1 - mads2) < 1e-8)
stopifnot(abs(mean(x$get_scales()$medians)) < 1e-8)
lfqcopy <- lfqdata$get_copy()
lfqTrans <- lfqcopy$get_Transformer()
lfqTrans$log2()
#> Column added : log2_abundance
before <- lfqTrans$get_scales()
lfqTrans$robscale()
#> data is : TRUE
#> Joining with `by = join_by(sampleName, isotopeLabel, protein_Id, peptide_Id)`
after <- lfqTrans$get_scales()
stopifnot(abs(mean(before$medians) - mean(after$medians)) < 1e-8)
stopifnot(abs(mean(before$mads) - mean(after$mads)) < 1e-8)
# normalize data using vsn
lfqcopy <- lfqdata$get_copy()
lfqTrans <- lfqcopy$get_Transformer()
lfqTransCheck <- lfqcopy$get_Transformer()
lfqTransCheck$log2()
#> Column added : log2_abundance
lfqTransCheck$get_scales()
#> $medians
#> A_V1 A_V2 A_V3 A_V4 B_V1 B_V2 B_V3 B_V4
#> 4.463740 4.450835 4.444549 4.463231 4.537348 4.568077 4.495236 4.519219
#> Ctrl_V1 Ctrl_V2 Ctrl_V3 Ctrl_V4
#> 4.500299 4.453443 4.491344 4.464994
#>
#> $mads
#> A_V1 A_V2 A_V3 A_V4 B_V1 B_V2 B_V3 B_V4
#> 0.3515227 0.3699336 0.3679643 0.3853720 0.3884622 0.3747087 0.3602410 0.3672785
#> Ctrl_V1 Ctrl_V2 Ctrl_V3 Ctrl_V4
#> 0.3507487 0.3606693 0.3632381 0.3691903
#>
lfqTransCheck$lfq$get_Plotter()$intensity_distribution_density()
#> Warning: Removed 225 rows containing non-finite outside the scale range
#> (`stat_density()`).
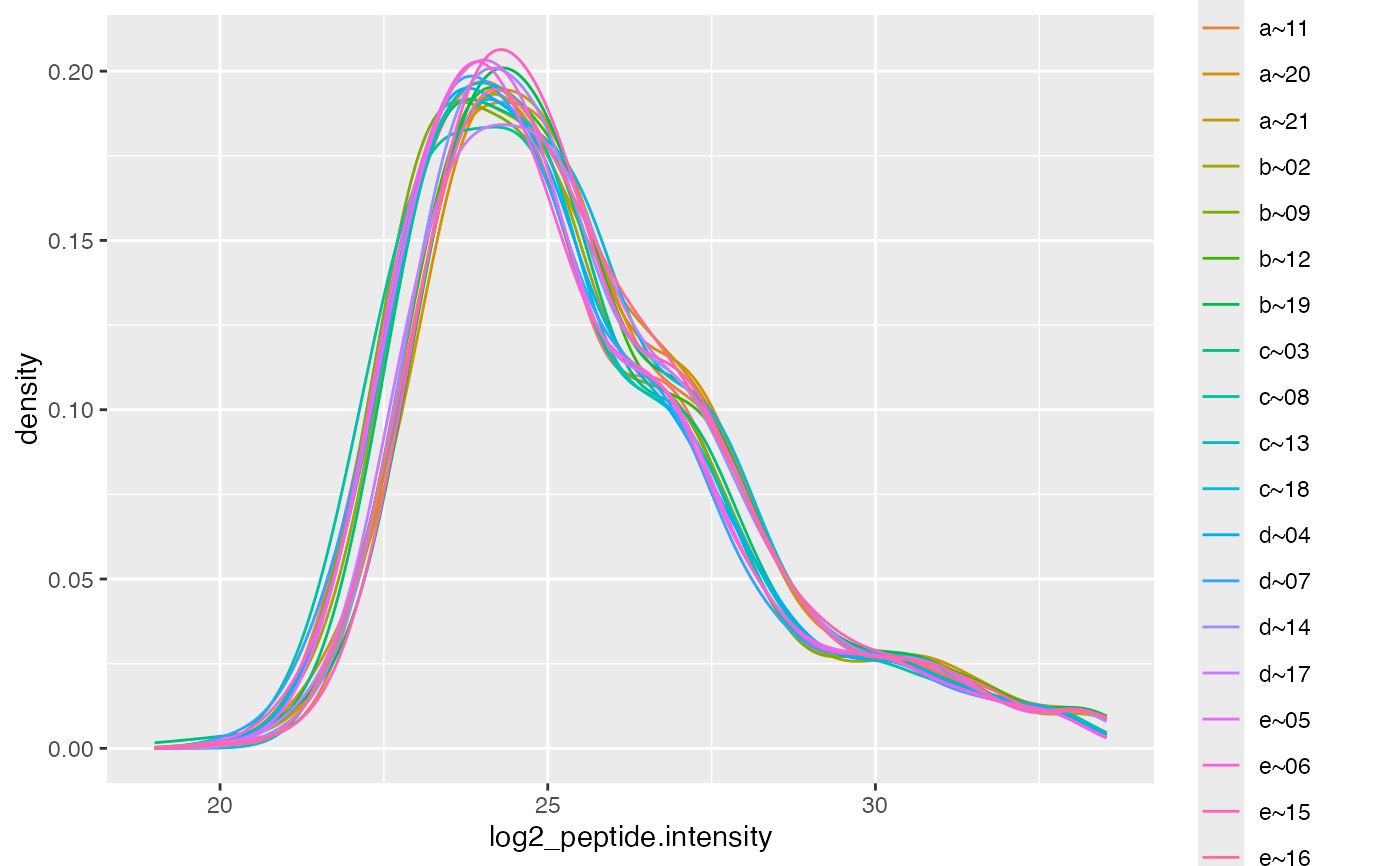 if(require("vsn")){
res <- lfqTrans$intensity_matrix( .func = vsn::justvsn)
res$lfq$get_Plotter()$intensity_distribution_density()
res$get_scales()
}
#> Loading required package: vsn
#> Joining with `by = join_by(sampleName, isotopeLabel, protein_Id, peptide_Id)`
#> $medians
#> A_V1 A_V2 A_V3 A_V4 B_V1 B_V2 B_V3 B_V4
#> 5.057603 5.050276 5.042556 5.053200 5.097080 5.119346 5.079693 5.090355
#> Ctrl_V1 Ctrl_V2 Ctrl_V3 Ctrl_V4
#> 5.089038 5.057453 5.079671 5.062745
#>
#> $mads
#> A_V1 A_V2 A_V3 A_V4 B_V1 B_V2 B_V3 B_V4
#> 0.2370482 0.2559391 0.2406615 0.2473880 0.2620343 0.2563651 0.2238271 0.2357968
#> Ctrl_V1 Ctrl_V2 Ctrl_V3 Ctrl_V4
#> 0.2646918 0.2331200 0.2431564 0.2435988
#>
if(require("preprocessCore")){
quant <- function(y){
ynorm <- preprocessCore::normalize.quantiles(y)
rownames(ynorm) <- rownames(y)
colnames(ynorm) <- colnames(y)
return(ynorm)
}
res <- lfqTrans$intensity_matrix( .func = quant)
res$lfq$get_Plotter()$intensity_distribution_density()
}
#> Loading required package: preprocessCore
#> Warning: data already transformed. If you still want to log2 tranform, set force = TRUE
#> Warning: Removed 225 rows containing non-finite outside the scale range
#> (`stat_density()`).
if(require("vsn")){
res <- lfqTrans$intensity_matrix( .func = vsn::justvsn)
res$lfq$get_Plotter()$intensity_distribution_density()
res$get_scales()
}
#> Loading required package: vsn
#> Joining with `by = join_by(sampleName, isotopeLabel, protein_Id, peptide_Id)`
#> $medians
#> A_V1 A_V2 A_V3 A_V4 B_V1 B_V2 B_V3 B_V4
#> 5.057603 5.050276 5.042556 5.053200 5.097080 5.119346 5.079693 5.090355
#> Ctrl_V1 Ctrl_V2 Ctrl_V3 Ctrl_V4
#> 5.089038 5.057453 5.079671 5.062745
#>
#> $mads
#> A_V1 A_V2 A_V3 A_V4 B_V1 B_V2 B_V3 B_V4
#> 0.2370482 0.2559391 0.2406615 0.2473880 0.2620343 0.2563651 0.2238271 0.2357968
#> Ctrl_V1 Ctrl_V2 Ctrl_V3 Ctrl_V4
#> 0.2646918 0.2331200 0.2431564 0.2435988
#>
if(require("preprocessCore")){
quant <- function(y){
ynorm <- preprocessCore::normalize.quantiles(y)
rownames(ynorm) <- rownames(y)
colnames(ynorm) <- colnames(y)
return(ynorm)
}
res <- lfqTrans$intensity_matrix( .func = quant)
res$lfq$get_Plotter()$intensity_distribution_density()
}
#> Loading required package: preprocessCore
#> Warning: data already transformed. If you still want to log2 tranform, set force = TRUE
#> Warning: Removed 225 rows containing non-finite outside the scale range
#> (`stat_density()`).
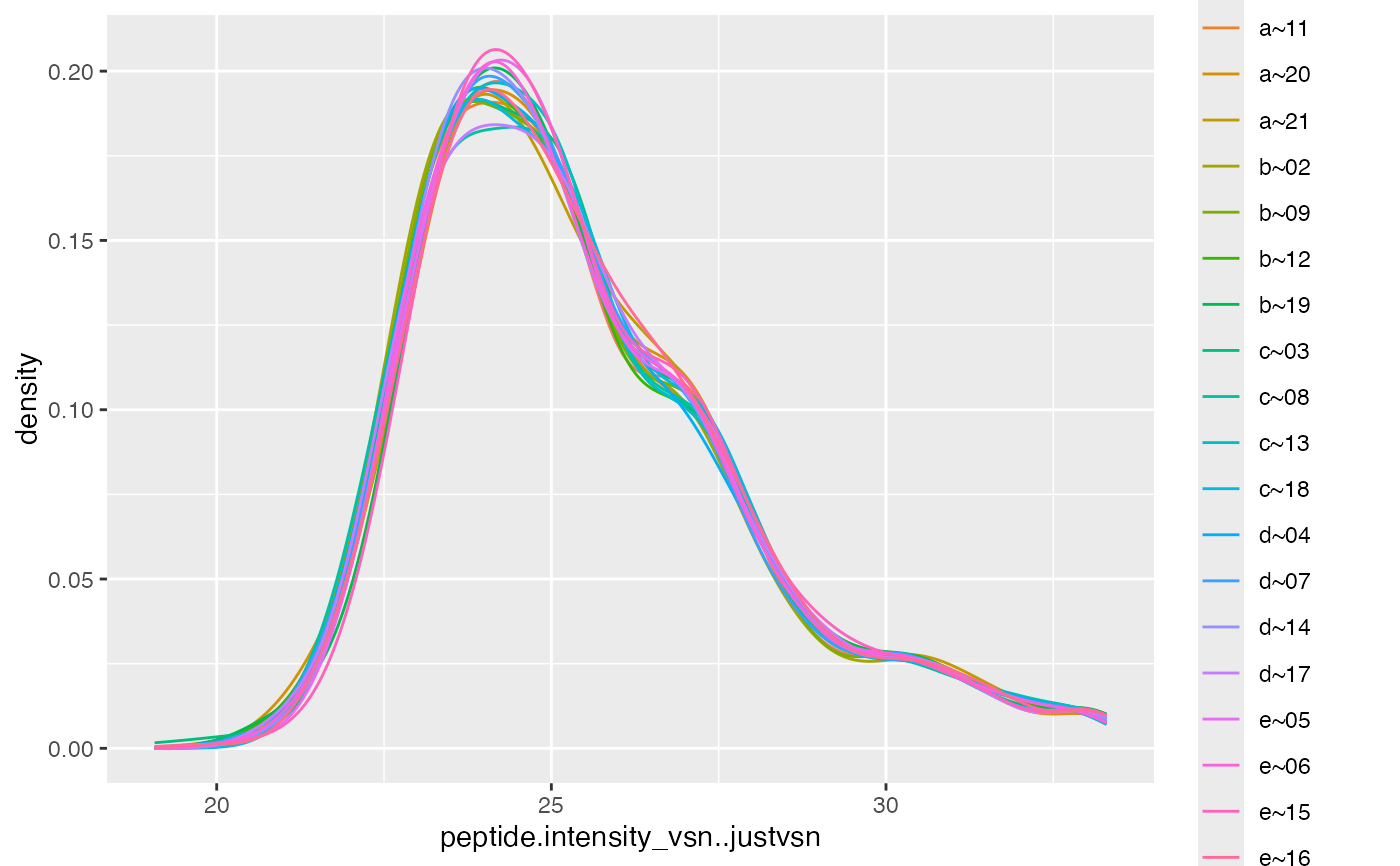 #subset scaling
istar2 <- sim_lfq_data_peptide_config()
#> creating sampleName from file_name column
#> completing cases
#> completing cases done
#> setup done
lfqdata2 <- LFQData$new(istar2$data, istar2$config)
lfqdata2 <- lfqdata2$get_Transformer()$intensity_array(log2)$lfq
#> Column added : log2_abundance
head(lfqdata2$hierarchy())
#> # A tibble: 6 × 1
#> protein_Id
#> <chr>
#> 1 0EfVhX~0087
#> 2 7cbcrd~5725
#> 3 9VUkAq~4703
#> 4 BEJI92~5282
#> 5 CGzoYe~2147
#> 6 DoWup2~5896
internal <- lfqdata2$get_subset(head(lfqdata2$hierarchy()))
#> Joining with `by = join_by(protein_Id)`
internal$hierarchy()
#> # A tibble: 6 × 1
#> protein_Id
#> <chr>
#> 1 0EfVhX~0087
#> 2 7cbcrd~5725
#> 3 9VUkAq~4703
#> 4 BEJI92~5282
#> 5 CGzoYe~2147
#> 6 DoWup2~5896
tr <- lfqdata2$get_Transformer()
tr$center_to_reference(internal)
#> data is transformed: TRUE
pl <- tr$lfq$get_Plotter()
pl$intensity_distribution_density()
#> Warning: Removed 36 rows containing non-finite outside the scale range
#> (`stat_density()`).
#subset scaling
istar2 <- sim_lfq_data_peptide_config()
#> creating sampleName from file_name column
#> completing cases
#> completing cases done
#> setup done
lfqdata2 <- LFQData$new(istar2$data, istar2$config)
lfqdata2 <- lfqdata2$get_Transformer()$intensity_array(log2)$lfq
#> Column added : log2_abundance
head(lfqdata2$hierarchy())
#> # A tibble: 6 × 1
#> protein_Id
#> <chr>
#> 1 0EfVhX~0087
#> 2 7cbcrd~5725
#> 3 9VUkAq~4703
#> 4 BEJI92~5282
#> 5 CGzoYe~2147
#> 6 DoWup2~5896
internal <- lfqdata2$get_subset(head(lfqdata2$hierarchy()))
#> Joining with `by = join_by(protein_Id)`
internal$hierarchy()
#> # A tibble: 6 × 1
#> protein_Id
#> <chr>
#> 1 0EfVhX~0087
#> 2 7cbcrd~5725
#> 3 9VUkAq~4703
#> 4 BEJI92~5282
#> 5 CGzoYe~2147
#> 6 DoWup2~5896
tr <- lfqdata2$get_Transformer()
tr$center_to_reference(internal)
#> data is transformed: TRUE
pl <- tr$lfq$get_Plotter()
pl$intensity_distribution_density()
#> Warning: Removed 36 rows containing non-finite outside the scale range
#> (`stat_density()`).
 lfqdata2$get_Plotter()$intensity_distribution_density()
#> Warning: Removed 36 rows containing non-finite outside the scale range
#> (`stat_density()`).
lfqdata2$get_Plotter()$intensity_distribution_density()
#> Warning: Removed 36 rows containing non-finite outside the scale range
#> (`stat_density()`).
 robscale <- lfqdata2$get_Transformer()$robscale_subset(internal)$lfq
#> data is : TRUE
#> Joining with `by = join_by(sampleName, isotopeLabel, protein_Id, peptide_Id)`
robscale$get_Plotter()$intensity_distribution_density()
#> Warning: Removed 36 rows containing non-finite outside the scale range
#> (`stat_density()`).
robscale <- lfqdata2$get_Transformer()$robscale_subset(internal)$lfq
#> data is : TRUE
#> Joining with `by = join_by(sampleName, isotopeLabel, protein_Id, peptide_Id)`
robscale$get_Plotter()$intensity_distribution_density()
#> Warning: Removed 36 rows containing non-finite outside the scale range
#> (`stat_density()`).