Limma moderated contrasts
Limma moderated contrasts
See also
Other modelling:
AnovaExtractor,
Contrasts,
ContrastsDEqMSFacade,
ContrastsDEqMSVoomFacade,
ContrastsFacadeBase,
ContrastsFirth,
ContrastsFirthFacade,
ContrastsFirthNestedFacade,
ContrastsLMFacade,
ContrastsLMImputeFacade,
ContrastsLMMissingFacade,
ContrastsLimma,
ContrastsLimmaFacade,
ContrastsLimmaImputeFacade,
ContrastsLimmaVoomFacade,
ContrastsLimmaVoomImputeFacade,
ContrastsLimpaFacade,
ContrastsLimpaNestedFacade,
ContrastsLmerNestedFacade,
ContrastsMissing,
ContrastsModeratedDEqMS,
ContrastsPlotter,
ContrastsRLMFacade,
ContrastsROPECA,
ContrastsROPECANestedFacade,
ContrastsRfitFacade,
ContrastsRfitImputeFacade,
ContrastsTable,
INTERNAL_FUNCTIONS_BY_FAMILY,
LR_test(),
Model,
ModelFirth,
ModelLimma,
StrategyLM,
StrategyLimma,
StrategyLimpa,
StrategyLmer,
StrategyLogistf,
StrategyRLM,
StrategyRfit,
build_contrast_analysis(),
build_model(),
build_model_glm_peptide(),
build_model_glm_protein(),
build_model_impute(),
build_model_limma(),
build_model_limma_impute(),
build_model_limma_voom(),
build_model_limma_voom_impute(),
build_model_limpa(),
build_model_logistf(),
compute_borrowed_variance(),
compute_borrowed_variance_limma(),
compute_contrast(),
compute_lmer_contrast(),
contrasts_fisher_exact(),
df.residual.rfit_prolfqua(),
get_anova_df(),
get_complete_model_fit(),
get_p_values_pbeta(),
group_label(),
impute_refit_singular(),
is_singular_lm(),
linfct_all_possible_contrasts(),
linfct_factors_contrasts(),
linfct_from_model(),
linfct_matrix_contrasts(),
list_facades(),
lookup_facade(),
merge_contrasts_results(),
model_analyse(),
model_summary(),
moderated_p_deqms(),
moderated_p_deqms_long(),
moderated_p_limma(),
moderated_p_limma_long(),
new_imputed_model(),
pivot_model_contrasts_to_wide(),
plot_lmer_peptide_predictions(),
register_facade(),
sigma.rfit_prolfqua(),
sim_build_models_lm(),
sim_build_models_lmer(),
sim_build_models_logistf(),
sim_make_model_lm(),
sim_make_model_lmer(),
strategy_limma(),
strategy_limpa(),
strategy_logistf(),
summary_ROPECA_median_p.scaled(),
unregister_facade(),
vcov.rfit_prolfqua()
Super class
prolfqua::ContrastsInterface -> ContrastsModerated
Public fields
ContrastClass implementing the Contrast interface
model_namename of model
subject_idcolumns with subject_id (proteinID)
p.adjustfunction to adjust p-values
variance_flooroptional lower bound for posterior variances
Methods
Inherited methods
prolfqua::ContrastsInterface$column_description()prolfqua::ContrastsInterface$contrast_summary_table()prolfqua::ContrastsInterface$extra_artifacts()prolfqua::ContrastsInterface$filter_significant()prolfqua::ContrastsInterface$get_config()prolfqua::ContrastsInterface$get_missing()prolfqua::ContrastsInterface$get_ora()prolfqua::ContrastsInterface$get_rank()
Method new()
initialize
Usage
ContrastsModerated$new(
Contrast,
model_name = paste0(Contrast$model_name, "_moderated"),
p.adjust = prolfqua::adjust_p_values,
variance_floor = NULL
)Method get_Plotter()
get ContrastsPlotter
Method to_wide()
convert to wide format
Usage
ContrastsModerated$to_wide(columns = c("p.value", "FDR", "statistic"))Examples
istar <- sim_lfq_data_protein_config(Nprot = 50)
#> creating sampleName from file_name column
#> completing cases
#> completing cases done
#> setup done
protIntensity <- istar$data
config <- istar$config
lProt <- LFQData$new(protIntensity, config)
lProt$rename_response("transformedIntensity")
modelFunction <-
strategy_lm("transformedIntensity ~ group_")
mod <- build_model(
lProt,
modelFunction)
Contr <- c("dil.b_vs_a" = "group_A - group_Ctrl")
contrast <- prolfqua::Contrasts$new(mod,
Contr)
contrast <- ContrastsModerated$new(contrast)
bb <- contrast$get_contrasts()
#> determine linear functions:
#> get_contrasts -> contrasts_linfct
#> contrasts_linfct
#> Joining with `by = join_by(protein_Id, contrast)`
csi <- ContrastsMissing$new(lProt, contrasts = Contr)
#> Warning: ContrastsMissing is deprecated: it substitutes group means rather than fitting a model. Prefer build_model_impute (LOD-imputed per-protein refit with borrowed variance) via the lm_impute / limma_impute facades. See ?ContrastsMissing for details.
contrast$get_contrasts() |> dim()
#> [1] 49 14
(xx <- csi$get_contrasts()) |> dim()
#> dil.b_vs_a=group_A - group_Ctrl
#> dil.b_vs_a=group_A - group_Ctrl
#> dil.b_vs_a=group_A - group_Ctrl
#> [1] 50 21
merged <- merge_contrasts_results(contrast, csi)
#> Joining with `by = join_by(protein_Id, contrast)`
#> Joining with `by = join_by(protein_Id, contrast)`
merged$more$get_contrasts() |> dim()
#> [1] 1 14
stopifnot(all(dim(merged$merged$get_contrasts()) == c(50, 14)))
stopifnot(all(dim(merged$same$get_contrasts()) == c(49, 14)))
cs <- contrast$get_contrast_sides()
cslf <- contrast$get_linfct()
ctr <- contrast$get_contrasts()
ctrwide <- contrast$to_wide()
cp <- contrast$get_Plotter()
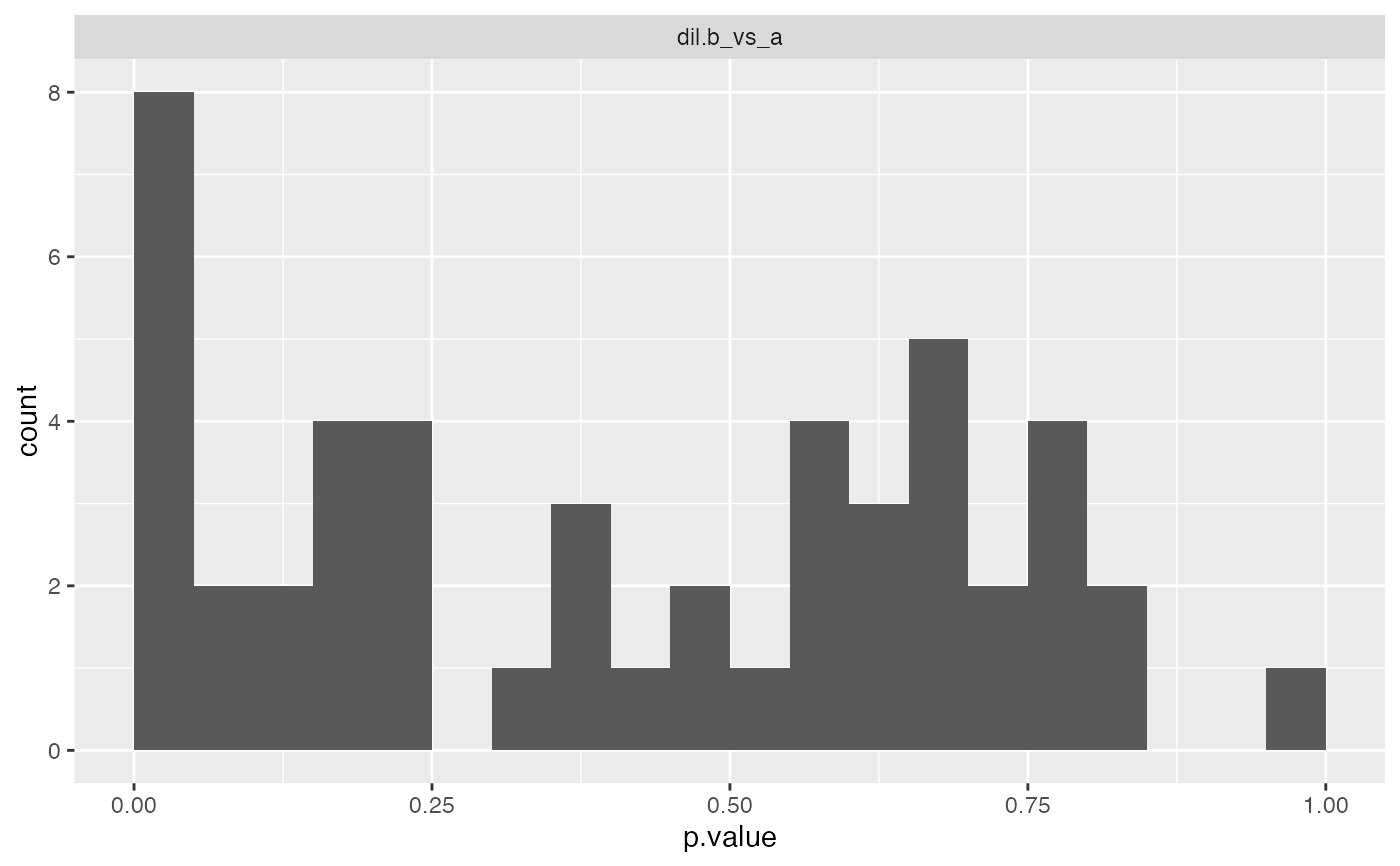
print(cp$histogram()$p.value, vp=NULL)
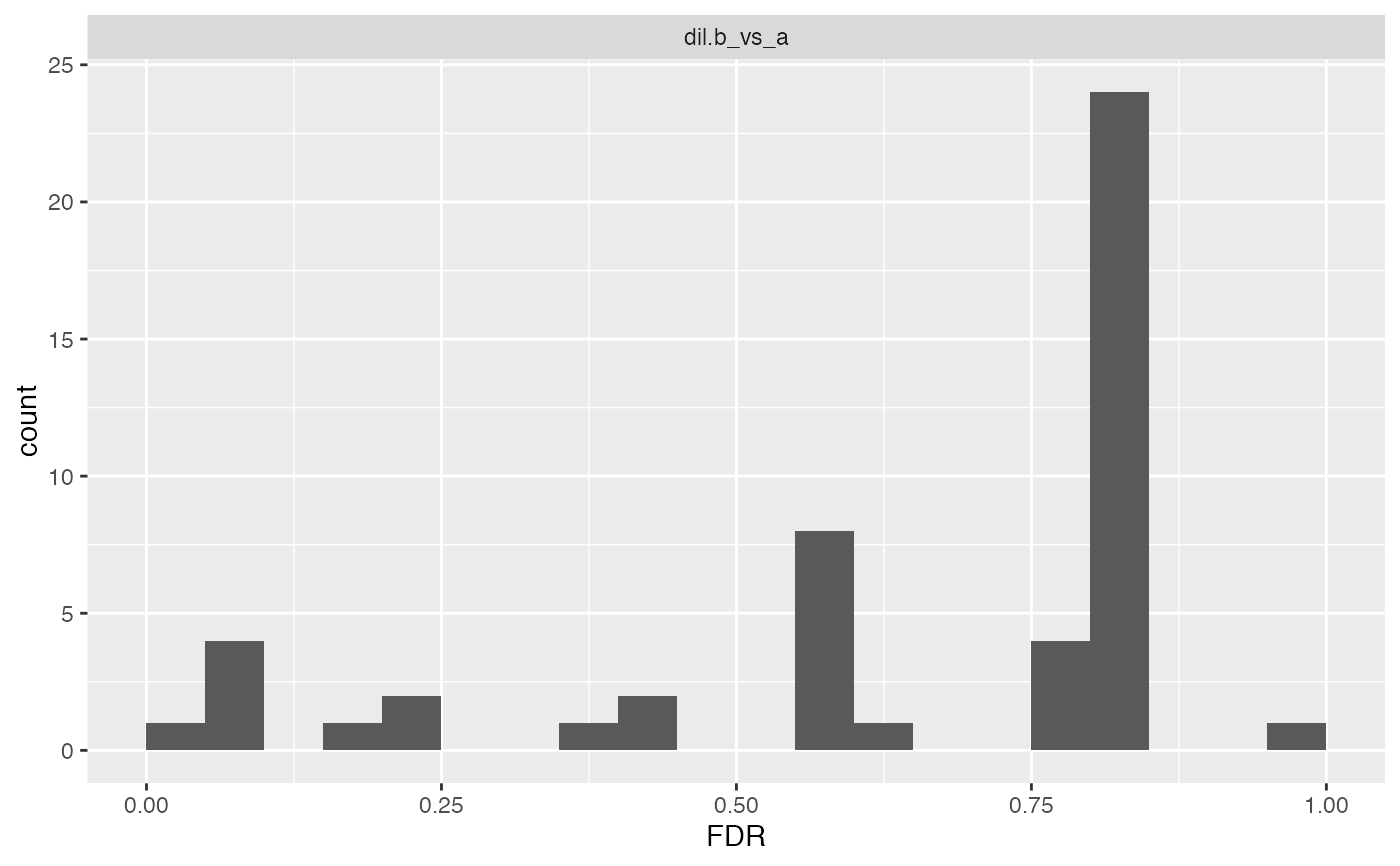
 print(cp$histogram()$FDR, vp = NULL)
print(cp$histogram()$FDR, vp = NULL)
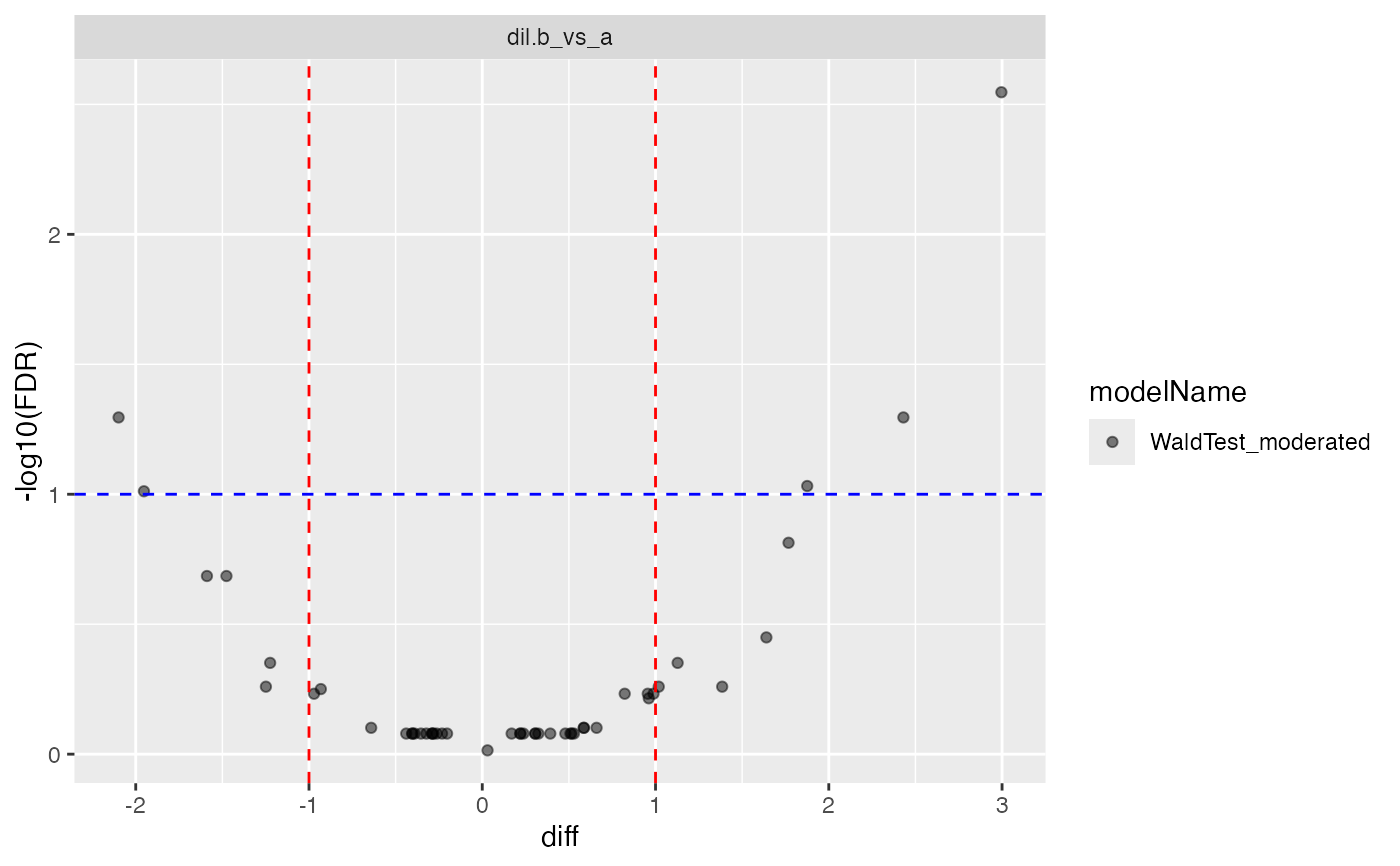
 cp$volcano()
#> $FDR
cp$volcano()
#> $FDR
 #>
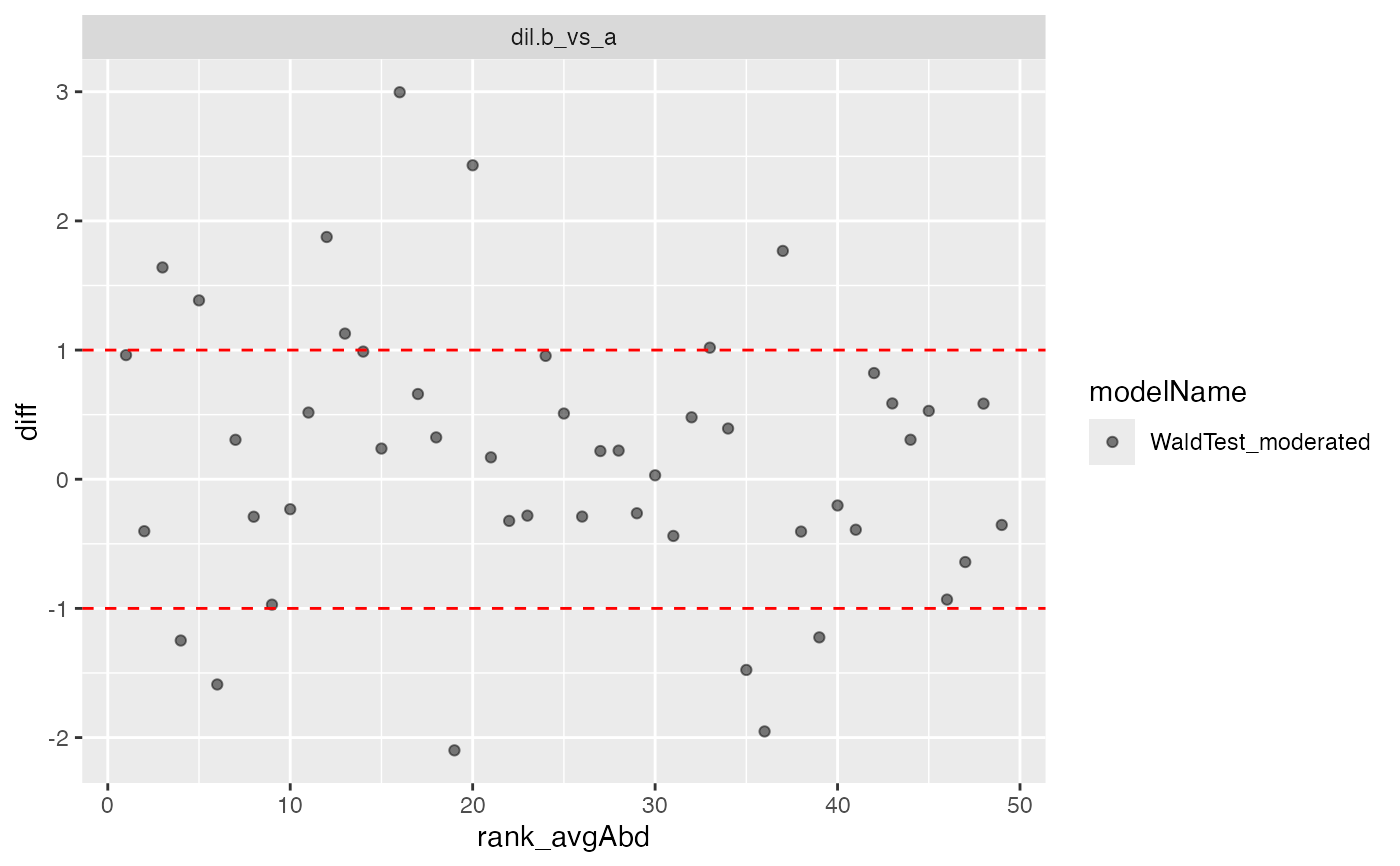
cp$ma_plot()
#>
cp$ma_plot()