holds results when contrasts are added.
holds results when contrasts are added.
See also
summary_ROPECA_median_p.scaled
Other modelling:
AnovaExtractor,
Contrasts,
ContrastsDEqMSFacade,
ContrastsDEqMSVoomFacade,
ContrastsFacadeBase,
ContrastsFirth,
ContrastsFirthFacade,
ContrastsFirthNestedFacade,
ContrastsLMFacade,
ContrastsLMImputeFacade,
ContrastsLMMissingFacade,
ContrastsLimma,
ContrastsLimmaFacade,
ContrastsLimmaImputeFacade,
ContrastsLimmaVoomFacade,
ContrastsLimmaVoomImputeFacade,
ContrastsLimpaFacade,
ContrastsLimpaNestedFacade,
ContrastsLmerNestedFacade,
ContrastsMissing,
ContrastsModerated,
ContrastsModeratedDEqMS,
ContrastsPlotter,
ContrastsRLMFacade,
ContrastsROPECA,
ContrastsROPECANestedFacade,
ContrastsRfitFacade,
ContrastsRfitImputeFacade,
INTERNAL_FUNCTIONS_BY_FAMILY,
LR_test(),
Model,
ModelFirth,
ModelLimma,
StrategyLM,
StrategyLimma,
StrategyLimpa,
StrategyLmer,
StrategyLogistf,
StrategyRLM,
StrategyRfit,
build_contrast_analysis(),
build_model(),
build_model_glm_peptide(),
build_model_glm_protein(),
build_model_impute(),
build_model_limma(),
build_model_limma_impute(),
build_model_limma_voom(),
build_model_limma_voom_impute(),
build_model_limpa(),
build_model_logistf(),
compute_borrowed_variance(),
compute_borrowed_variance_limma(),
compute_contrast(),
compute_lmer_contrast(),
contrasts_fisher_exact(),
df.residual.rfit_prolfqua(),
get_anova_df(),
get_complete_model_fit(),
get_p_values_pbeta(),
group_label(),
impute_refit_singular(),
is_singular_lm(),
linfct_all_possible_contrasts(),
linfct_factors_contrasts(),
linfct_from_model(),
linfct_matrix_contrasts(),
list_facades(),
lookup_facade(),
merge_contrasts_results(),
model_analyse(),
model_summary(),
moderated_p_deqms(),
moderated_p_deqms_long(),
moderated_p_limma(),
moderated_p_limma_long(),
new_imputed_model(),
pivot_model_contrasts_to_wide(),
plot_lmer_peptide_predictions(),
register_facade(),
sigma.rfit_prolfqua(),
sim_build_models_lm(),
sim_build_models_lmer(),
sim_build_models_logistf(),
sim_make_model_lm(),
sim_make_model_lmer(),
strategy_limma(),
strategy_limpa(),
strategy_logistf(),
summary_ROPECA_median_p.scaled(),
unregister_facade(),
vcov.rfit_prolfqua()
Super class
prolfqua::ContrastsInterface -> ContrastsTable
Methods
Inherited methods
prolfqua::ContrastsInterface$column_description()prolfqua::ContrastsInterface$contrast_summary_table()prolfqua::ContrastsInterface$extra_artifacts()prolfqua::ContrastsInterface$filter_significant()prolfqua::ContrastsInterface$get_config()prolfqua::ContrastsInterface$get_missing()prolfqua::ContrastsInterface$get_ora()prolfqua::ContrastsInterface$get_rank()
Method new()
intitialize
Usage
ContrastsTable$new(
contrastsdf,
subject_id = "protein_Id",
model_name = "ContrastTable"
)Method to_wide()
convert to wide format
Usage
ContrastsTable$to_wide(columns = c("p.value", "FDR", "statistic"))Examples
bb <-prolfqua::sim_lfq_data_peptide_config()
#> creating sampleName from file_name column
#> completing cases
#> completing cases done
#> setup done
configur <- bb$config$clone(deep=TRUE)
configur$hierarchy_depth <- 2
data <- bb$data
lfqdata <- LFQData$new(data, configur)
lfqdata$factors()
#> # A tibble: 12 × 3
#> sample sampleName group_
#> <chr> <chr> <chr>
#> 1 A_V1 A_V1 A
#> 2 A_V2 A_V2 A
#> 3 A_V3 A_V3 A
#> 4 A_V4 A_V4 A
#> 5 B_V1 B_V1 B
#> 6 B_V2 B_V2 B
#> 7 B_V3 B_V3 B
#> 8 B_V4 B_V4 B
#> 9 Ctrl_V1 Ctrl_V1 Ctrl
#> 10 Ctrl_V2 Ctrl_V2 Ctrl
#> 11 Ctrl_V3 Ctrl_V3 Ctrl
#> 12 Ctrl_V4 Ctrl_V4 Ctrl
Contrasts <- c("aC" = "group_A - group_Ctrl",
"bC" = "group_A - group_Ctrl")
csi <- ContrastsMissing$new(lfqdata, contrasts = Contrasts)
#> Warning: ContrastsMissing is deprecated: it substitutes group means rather than fitting a model. Prefer build_model_impute (LOD-imputed per-protein refit with borrowed variance) via the lm_impute / limma_impute facades. See ?ContrastsMissing for details.
ctr <- csi$get_contrasts()
#> aC=group_A - group_Ctrl
#> bC=group_A - group_Ctrl
#> aC=group_A - group_Ctrl
#> bC=group_A - group_Ctrl
#> aC=group_A - group_Ctrl
#> bC=group_A - group_Ctrl
csi$subject_id
#> [1] "protein_Id" "peptide_Id"
xcx <- ContrastsTable$new(ctr, subject_id = csi$subject_id, model_name = "TableTest")
xcx$get_contrasts()
#> # A tibble: 56 × 22
#> modelName estimate_type protein_Id peptide_Id meanAbundanceImp_group_1
#> <chr> <chr> <chr> <chr> <dbl>
#> 1 groupAverage missing_fallback 0EfVhX~0087 ITLb4x1q 18.0
#> 2 groupAverage missing_fallback 0EfVhX~0087 ahQLlQY7 25.8
#> 3 groupAverage missing_fallback 0EfVhX~0087 dJkdz7so 15.5
#> 4 groupAverage missing_fallback 7cbcrd~5725 D5dQ4nKk 29.5
#> 5 groupAverage missing_fallback 9VUkAq~4703 eIC06D7g 18.0
#> 6 groupAverage missing_fallback BEJI92~5282 HBkZvdhT 16.8
#> 7 groupAverage missing_fallback BEJI92~5282 qQ1GK8Un 23.8
#> 8 groupAverage missing_fallback CGzoYe~2147 mjHSHhoe 24.3
#> 9 groupAverage missing_fallback DoWup2~5896 KVUnZ6oZ 23.8
#> 10 groupAverage missing_fallback Fl4JiV~8625 GsUIOl6Q 19.1
#> # ℹ 46 more rows
#> # ℹ 17 more variables: meanAbundanceImp_group_2 <dbl>, diff <dbl>,
#> # group_1_name <chr>, group_2_name <chr>, contrast <chr>, avgAbd <dbl>,
#> # indic <dbl>, nrMeasured_group_1 <int>, nrMeasured_group_2 <int>, df <int>,
#> # sigma <dbl>, std.error <dbl>, statistic <dbl>, p.value <dbl>,
#> # conf.low <dbl>, conf.high <dbl>, FDR <dbl>
xcx$get_Plotter()$volcano()
#> $p.value
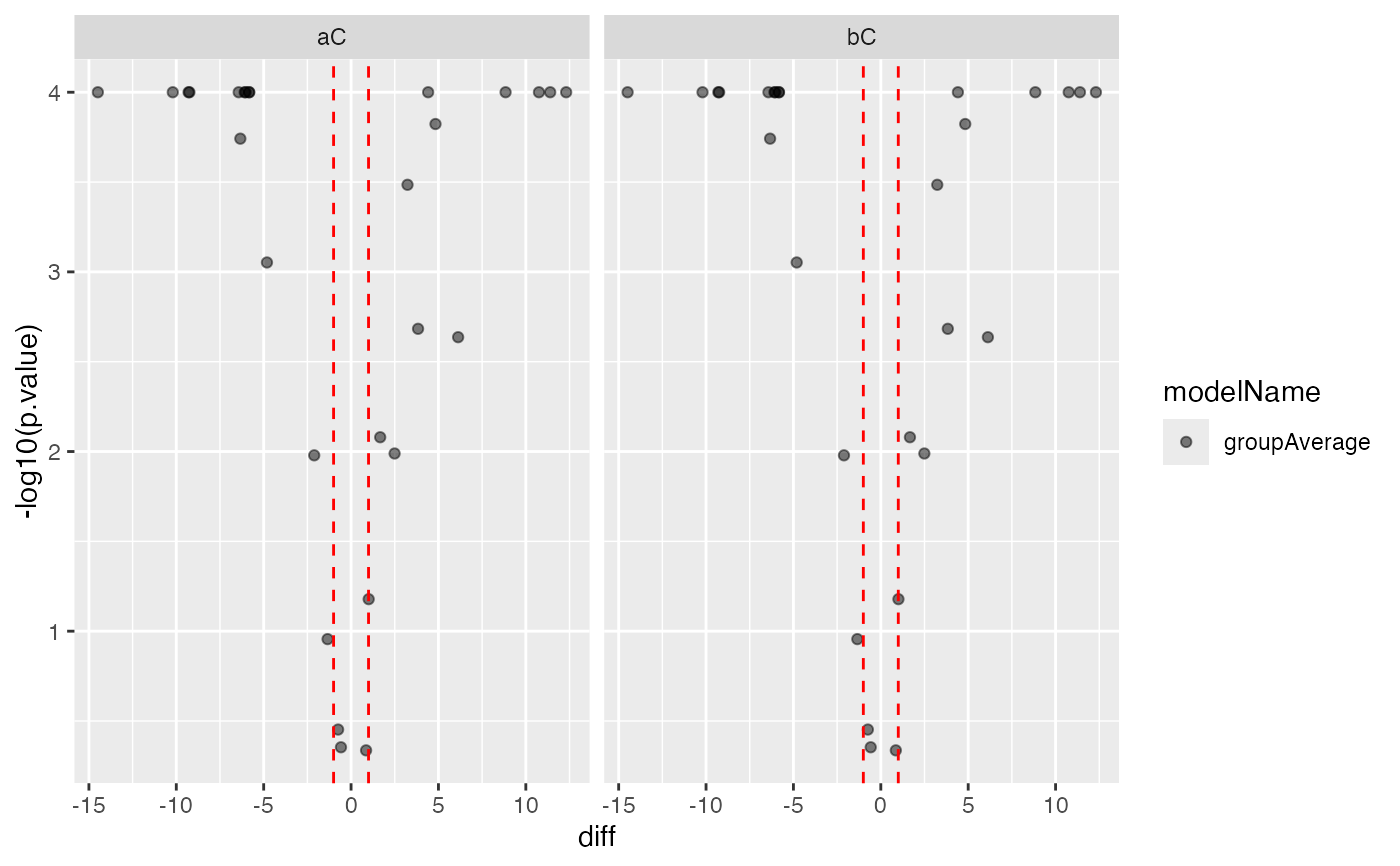 #>
#> $FDR
#>
#> $FDR
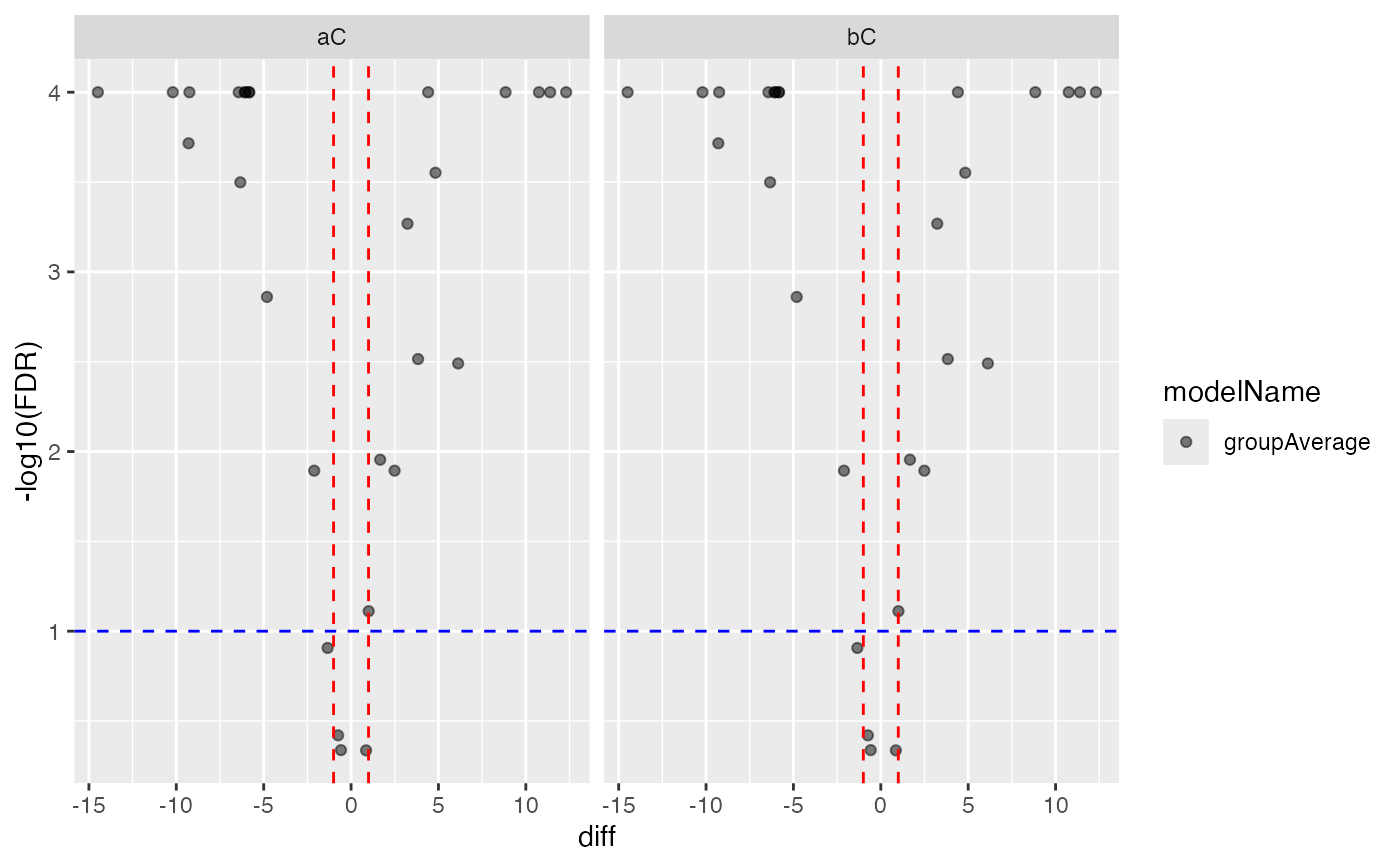 #>
stopifnot(is.null(xcx$get_contrast_sides()))
stopifnot(is.null(xcx$get_linfct()))
stopifnot(ncol(xcx$to_wide()) == 10)
#>
stopifnot(is.null(xcx$get_contrast_sides()))
stopifnot(is.null(xcx$get_linfct()))
stopifnot(ncol(xcx$to_wide()) == 10)